通常、重度免疫能低下を呈することはないとされていた18q欠失症候群
東京医科歯科大学は6月25日、成人期になって易感染性を示した18q欠失症候群の患者で、遅発性複合免疫不全症を生じていることを初めて明らかにしたと発表した。この研究は、同大大学院医歯学総合研究科小児地域成育医療学講座の金兼弘和寄付講座教授、同大発生発達病態学分野の友政弾大学院生ら、鹿児島大学大学院医歯学総合研究科小児科学分野の西川拓朗准教授、岡本康裕教授ら、やまびこ医療福祉センター(鹿児島市)、広島大学の研究グループによるもの。研究成果は、「Journal of Clinical Immunology」に掲載されている。
画像はリリースより
(詳細は▼関連リンクからご確認ください)
18q欠失症候群は、18番染色体長腕の一部分が失われること(欠失)を原因とする先天性の染色体異常症。4~5.5万出生に1人程度の発症とされている。染色体欠失部位に存在していた、いくつかの遺伝子の機能が失われることが症状の原因になると考えられる。症状としては、精神運動発達遅滞、先天性大脳白質形成不全症、特徴的な顔立ち(顔の正中の低形成)、筋緊張低下、低身長、目の異常、難聴、口蓋裂、骨格の異常、心疾患等が見られる。18q欠失症候群の根本的な治療法はないため、それぞれの症状に応じた対症療法が行われている。生命予後は重症感染症や心疾患などの合併症がなければ、良好とされる。また、18q欠失症候群患者は、先天性免疫異常症(inborn errors of immunity:IEI)の一つでもあり、50~90%の患者で1種類以上の免疫グロブリンの低下があり、液性免疫不全症を呈する。しかし、通常、それ以上の重度の免疫能低下を呈することはないとされていた。
ニューモシスチス肺炎発症の28歳男性/多発リンパ節腫大と肺炎発症の48歳女性
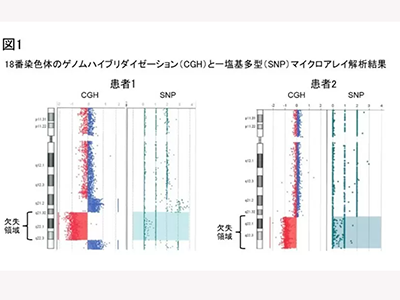
これまで感染症罹患がほとんどなかった18q欠失症候群である28歳男性(患者1)が、ニューモシスチス肺炎を発症。もう一人の患者(患者2)は48歳女性で多発リンパ節腫大と肺炎の精査の過程の染色体検査で、18q欠失を認めた。今回の研究では、アレイベースゲノムハイブリダイゼーション解析で両患者の精査を実施。その結果、患者1では18q21.32-q22.3に、患者2では18q21.33-qterに欠失を認めた。
両患者は液性/細胞性免疫不全症を合併、LOCIDと診断
2人の患者の免疫状態を調べたところ、低ガンマグロブリン血症、メモリーB細胞とナイーブCD4+および/またはCD8+細胞数の減少、CFSEによるT細胞分裂試験での反応低下、T細胞受容体のレパトア解析での偏り、T細胞受容体遺伝子再構成断片(TREC:T-cell receptor recombination excision circles)およびIgκ鎖遺伝子再構成断片(KREC:Igκ-deleting recombination excision circles)の低レベルが認められた。それらの結果から、両患者は液性免疫不全症のみなならず細胞性免疫不全症を合併しており、両患者は遅発性複合免疫不全症(LOCID:ate-onset combined immunodeficiency)と診断された。そして、両患者において、全エクソーム解析やターゲットシーケンス等の遺伝子解析を追加したが、18q欠失症候群以外にLOCIDを生じるような遺伝子変異は認められなかった。
18q欠失症候群患者、定期的に細胞性/液性免疫能検査を
18q欠失症候群の患者の一部は、低ガンマグロブリン血症を主とする液性免疫不全症を呈することが知られていた。今回の研究により、同疾患はさらに細胞性免疫不全症を合併し、より重症な遅発性の複合免疫不全症を引き起こすことがあることが明らかになった。今回初めての報告だったが、単に免疫状態がきちんと評価されていないだけの可能性がある。したがって、18q欠失症候群の患者は、定期的に細胞性/液性免疫能の検査を受けた方が良いと考えられる、と研究グループは述べている。
▼関連リンク
・東京医科歯科大学 プレスリリース


