多くのエピゲノム実験データ、活用するためには高度な解析技術と豊富な計算資源が必要
熊本大学は5月16日、エピゲノム統合データベース「ChIP-Atlas」のメジャーアップデートを行い、ゲノムの三次元構造・疾患感受性ゲノム変異などの注釈づけ情報を統合し、遺伝子発現制御に関わるエピゲノム状態の変容を検出する比較解析ツールを実装したと発表した。この研究は、同大生命資源研究・支援センターの鄒兆南助教、沖真弥教授、千葉大学国際高等研究基幹/大学院医学研究院の大田達郎准教授らの研究グループによるもの。研究成果は、「Nucleic Acids Research」にオンライン掲載されている。
画像はリリースより
(詳細は▼関連リンクからご確認ください)
ヒトの体の細胞はすべて同じゲノムを持つのに、なぜ細胞の種類ごとに異なる形をしていて、特異的な働きができるのか。その鍵を握っているのは、細胞タイプごとに特異な遺伝子セットを活性化する遺伝子発現制御機構である。ゲノム上に存在するエンハンサー領域の活性化、DNAのメチル化状態、そしてゲノムに結合する約1,000種類の転写因子が互いに協調して、DNAの塩基配列を変えることなく、遺伝子の機能を決めている。この仕組みのことを「エピゲノム」といい、十数年前に次世代シーケンサーが開発されて以来、このエピゲノムの仕組みを理解するために、さまざまなシーケンス技術が登場した。その中で、ゲノム上の転写因子結合場所を調べるChIP-seq、転写因子が結合しうる活性化エンハンサー領域を調べるATAC-seq、DNAのメチル化状態を調べるBisulfite-seqが広く利用されている。これまでに論文などで数十万件のエピゲノムに関する実験データが報告されてきたが、これらのデータの意味を理解して活用するためには、高度な解析技術と豊富な計算資源が必要だった。
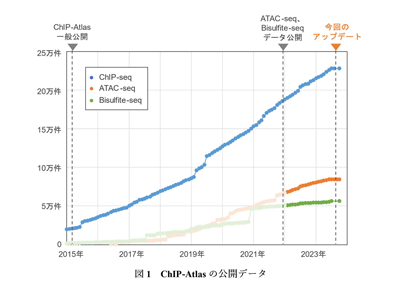
ヒト含む6モデル生物、40万件のChIP/ATAC/Bisulfite-seqデータ収集・計算
研究グループは、国立遺伝学研究所のスーパーコンピュータシステムを利用し、ヒト、マウス、ラット、ショウジョウバエ、線虫、出芽酵母の6つのモデル生物の約40万件のChIP-seq、ATAC-seq、Bisulfite-seqビッグデータを収集・計算した。
エピゲノム統合データベースChIP-Atlas、機能拡充で世界最大規模の地位を確立
これは、これらのシーケンス技術の解析可能な公共データをChIP-Atlasがほぼすべて網羅していることを意味する。ChIP-Atlasを利用することで、アトラス(地図帳)をめくるように自分の好きなゲノム領域にアクセスし、そこに結合する転写因子、活性化エンハンサー領域、DNAメチル化の情報をすべて閲覧できる。ChIP-Atlasのデータは、正常な細胞や組織だけでなく、遺伝子変異、薬物投与、加齢などによる変容も含んでおり、これらはホームページからキーワード検索で簡単に入手できる。これにより、ChIP-Atlasは世界最大規模のエピゲノム統合データベースという地位を確立した。そこで研究グループは、後述の機能拡充を含め、このたび正式に論文として報告した。
離れた位置のエンハンサー予測可能にするゲノムDNA三次元構造の実験データ統合
ChIP-Atlasによる活性化エンハンサー領域とDNAメチル化状態の可視化により、従来多大なコストを必要とされたエンハンサーの特定を、ウェブ上の簡単な操作で予測できるようになった。また、推定されたエンハンサー領域に結合する転写因子も視覚的に理解できるようになった。しかしながら、これだけではひとつひとつのエンハンサーがどの遺伝子の発現制御に関わるかを特定することができない。これは、ゲノムは三次元構造を形成しているため、エンハンサーはその最も近傍の遺伝子を標的としているとは限らないからである。例えば、手足の元となる肢芽の形成に重要なShh遺伝子のスイッチとなるエンハンサーは、Shh遺伝子から80万塩基以上離れたところに存在することが知られている。このような知見を可視化するため、研究グループはChIP-Atlasに「Annotation Track」として、さらにゲノムDNAの三次元構造を調べた実験データを統合した。
Annotation Trackの、利用事例は以下の通りである。例えば、ATAC-seqとChIP-seqデータから、乳がんの発生に寄与するエストロゲン受容体をコードするESR1遺伝子の周辺に複数の活性化エンハンサー領域が存在することを理解できるが、これだけではESR1の発現はどのエンハンサーによって調節されているかがわからない。しかしながら、この度のゲノム三次元構造データの統合により、ESR1はその上流約2万塩基のゲノム領域にコンタクトしていることがわかるようになった。つまり、ESR1は恐らくこの領域に位置するエンハンサーによる制御を受けるだろうと推察できる。
エンハンサー領域の疾患関連SNP情報も追加、発症メカニズムの推測も
さらに今回のアップデートでは、疾患に関連するゲノム上の一塩基多型(SNP)などの変異情報もAnnotation Trackに新たに追加された。これにより、エンハンサー領域の位置情報に加え、そこに存在する疾患関連SNPの情報を加味することで、疾患の背景にある遺伝子発現制御機構を一括で理解できるようになった。ESR1の上流に位置する活性化エンハンサー領域には、乳がんの発症リスク上昇に関連する複数SNPが報告されていることがわかる。このように、ウェブ上の簡単な操作のみで、ESR1遺伝子のエンハンサー領域には乳がんのSNPがあることが理解できる。このことからそのSNPがESR1のスイッチ役である転写因子群の結合異常をもたらす結果、エストロゲンの分泌異常が引き起こされるというメカニズムが推察できる。つまり、ChIP-Atlasの利活用により、ゲノム変異に起因する疾患の発症メカニズムに関するリーズナブルな仮説を形成することが可能である。
エピゲノム状態の変容、「Diff Analysis」で2群の実験データ比較解析も可能
前述の通り、ChIP-Atlasによるデータ統合により、ゲノム上の転写因子結合、活性化エンハンサー領域、DNAメチル化などのエピゲノム状態を反映した実験データを視覚的に理解できるようになった。また、このような可視化により、エピゲノム状態は細胞の分化段階、薬物を含む環境因子への曝露、疾患の発症などの影響を受け、ダイナミックに変化することがより鮮明にわかるようになった。つまり、これらの生体内イベントのメカニズムに迫るためには、その分子基盤であるエピゲノム状態の変容を正確に評価することが重要だが、これにもやはり情報学・統計学の知識と潤沢な計算資源が必要だった。
そこで、ChIP-AtlasではChIP-seq、ATAC-seq、Bisulfite-seqデータの比較解析を支援するオンライン解析ツール「Diff Analysis」を実装した。例えば多能性を有するES細胞と、筋肉の元となる筋芽細胞由来のATAC-seqデータを比較し、両者で活性化が異なるエンハンサー領域を簡単に抽出できる。例えば、細胞の多能性を規定するOct4遺伝子の周辺はES細胞で特異的に活性化しており、逆に筋肉のマーカー遺伝子であるMyod1の近傍には筋芽細胞特異的な活性化エンハンサー領域が存在することを視覚的に理解できる。このツールは、ChIP-Atlasにある数十万件の解析済み実験データによって成り立っている。これにより、ウェブ上で2群の実験データのアクセス番号を入力するだけで、わずか数分で計算が終了し比較結果を閲覧できるようになる。実験データの網羅性と操作の手軽さを兼ね備えたウェブサービスとしては世界初である。
遺伝性疾患メカニズム解明のほか、創薬標的発見や組織再生医療などの応用にも期待
ChIP-Atlasは2015年の一般公開以来、遺伝学、疾患メカニズム、創薬、発生生物学など幅広い研究分野で利用されており、これまでに1,000報に迫る論文に引用されている。この度のメジャーアップデートにより、疾患ゲノム情報をはじめとするゲノム・エピゲノム注釈づけデータが大幅に拡充され、ChIP-Atlasは遺伝子発現制御の異常に起因する遺伝性疾患の成り立ちを解明するデータ基盤へと進化した。
また、エピゲノムデータの比較解析ツールの実装は、薬物摂動によるエピゲノム状態の変容を突破口とした薬物の作用機序の解明、ひいては新規創薬標的の発見への応用が期待される。さらに、「細胞分化におけるエピゲノム変化に切り込むことで、例えば細胞の直接分化転換(いわゆるダイレクト・リプログラミング技術)を誘導できる司令塔的な転写因子もしくは低分子化合物をより簡便に見出すことが可能になる。将来的には組織再生医療への応用が期待される」と、研究グループは述べている。
▼関連リンク
・熊本大学 お知らせ


