再生医療法のもと「医療の安全性の確保」は本当に果たされているか
国立がん研究センターは11月20日、「再生医療等の安全性の確保等に関する法律(以下、再生医療法)」の現在までの施行状況および同法の改正の方向性を検討するため、同法で義務づけられる有害事象(疾病等)発生の報告状況を調査し、同法の目的である再生医療の安全性確保が果たされているか検討した結果、再生医療法に基づく自由診療において、有害事象の発生が適切に報告・検討されていない可能性が示唆されたことを発表した。この研究は、同センター生命倫理部の一家綱邦部長、静岡社会健康医学大学院大学八田太一講師、京都大学iPS細胞研究所、京都大学高等研究院ヒト生物学高等研究拠点の研究グループによるもの。研究成果は、「Stem Cell Reports」に掲載されている。
画像はリリースより
(詳細は▼関連リンクからご確認ください)
再生医療法は、日本における再生医療を国民が迅速かつ安全に受けられるようにするために、2013年に制定された。再生医療法に基づき、再生医療を「治療(現状の圧倒的多数は自由診療)」または「臨床研究」として実施する場合には、医療機関は厚生労働大臣の認定を受けた委員会に再生医療の提供計画を提出して審査を受けることになっている。医療に「絶対の安全性」はないからこそ、治療実施者は治療を実施した結果を把握し、検証することが求められる。そのための仕組みとして再生医療法は、有害事象(疾病等)の発生を認定再生医療等委員会に報告して審査を受けることを、再生医療実施者の義務としている。
これまでに研究グループは、自由診療で実施される再生医療、それを規制する再生医療法に関するさまざまな問題・課題・疑義を複数の論文で発表してきた。今回の論文は、「再生医療の安全性を確保することを目的とする法律は、本当に安全性を確保しているのだろうか?」という法律の重要なポイントを検証するために行った調査の結果をまとめたものになる。
研究より治療としての投与が桁違いに多い一方、有害事象報告件数は治療の方が少ない
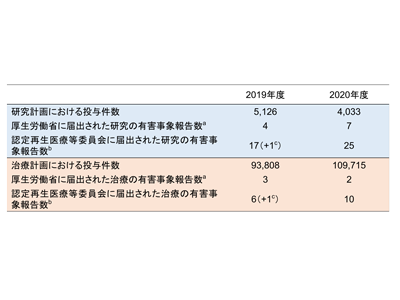
研究グループは、認定再生医療等委員会に公開義務のある委員会議事録(2019年度、2020年度分)を全て調べて、委員会で審議された疾病等報告の件数を算出した。それらの件数を、厚生労働省が毎年1回公表する日本で実施される全ての計画から報告された有害事象(疾病等)の件数「定期報告のとりまとめの概要」で公表されている「(細胞の)投与件数」と対比させた。その結果、治療計画における投与件数が研究計画における投与件数よりも桁違いに多いのにもかかわらず、有害事象(疾病等)の報告件数は治療計画の方が少ないことが明らかになった。具体的には、治療計画において、2019年度、2020年度ともに、およそ10万回の細胞投与に対して報告件数が10回未満に留まっていた。
再生医療等製品では3~4回の投与に対して1回の報告
研究グループは、この意味を検討するため、日本のもう1つの「再生医療の治療」といえる再生医療等製品の使用回数(投与件数)と有害事象の報告件数を調査した。再生医療等製品は、医薬品、医療機器等の品質、有効性および安全性の確保等に関する法律(薬機法)に基づいて、国が品質、安全性と有効性を認めたものに限って製造・販売を承認している。薬機法は、再生医療等製品の副作用・不具合等によるものと疑われる有害事象の発生について、当該製品の販売業者に厚生労働省に報告する義務を課している。
使用回数については、NDBオープンデータを使って推計した。ただし、調査対象とした製品は、最も正確な使用回数が推計できる「ジェイス」「ジャック」「テムセルHS注」の3つに限定した。有害事象の報告件数については、薬機法が施行された2014年11月25日以降、全ての再生医療等製品の使用に基づく「不具合等が疑われる症例報告」を公表している医薬品医療機器総合機構(PMDA)のデータから3製品について「患者等の健康被害状況」に記載がある症例報告を抽出した。
その結果、3つの再生医療等製品の使用においては、およそ3~4回の投与に対して1回の報告があることがわかった。また、重篤なものから軽微なものまでさまざまな有害事象の発生が報告されていた。
2つの法制度における有害事象報告の頻度の差、重大な結果
日本では2つの法制度に分かれて行われる「再生医療の治療」において、有害事象の報告頻度が大きく異なることは、再生医療法に基づく治療は必ずしも第三者性が担保されていない認定再生医療等委員会で審査されただけで実施されているのに対して、再生医療等製品は臨床試験の結果を国が慎重に検討して承認したものであることを踏まえると、重大な結果であると考えられる。
この結果からは、再生医療法に基づいて実施されている治療計画では、有害事象(疾病等)の報告制度が適切に機能しているのか、疑義が生じる。この疑義を裏付けるようなケース、すなわち、再生医療を受けた患者の有害事象発生についての訴えが、実施者に受け入れられず、その計画を審査した委員会に報告されていないケースがあることも、過去のメディア報道と今回の調査から推測された。
さらに、2つの法制度における有害事象報告の頻度の違いは、両法制度に携わる関係者の「医療における安全性の確保」に対する意識の違いの表れであるとも考えられる。有害事象の報告制度が適切に機能していないとしたら、「安全性を確保する」という再生医療法の目的が達成されているか否かを検証することはできないとも考えられる。
奇しくも、最近、国際細胞・遺伝子治療学会(International Society for Cell & Gene Therapy)によって、各国が有害事象の報告制度を見直し、その報告データを活用すること、そのような取り組みの前提として患者からの報告制度を設けることが提案された。再生医療法の改正においては、こうした提案を真摯に検討することが望ましいと考えられる。
「現在進められている再生医療法の改正の議論に社会的な関心が向けられ、結果的に再生医療法の改正や現行制度の運用の見直しがより適切なものになること、また、患者さんの再生医療に対する理解や治療選択の参考になることを願っている」と、研究グループは述べている。
▼関連リンク
・国立がん研究センター プレスリリース

