孤発性/家族性PDと関わりがあるCHCHD2の詳細は?
東京医科歯科大学は8月14日、PARK22/CHCHD2変異によるパーキンソン病の発症メカニズムを明らかにしたと発表した。この研究は、同大難治疾患研究所・病態細胞生物分野の鳥居暁プロジェクト准教授、清水重臣教授、順天堂大学大学院医学研究科神経学の佐藤栄人先任准教授、服部信孝教授らの研究グループによるもの。研究成果は、「EMBO Molecular Medicine」に掲載されている。
画像はリリースより
(詳細は▼関連リンクからご確認ください)
パーキンソン病の発症には、中脳黒質ドーパミン作動性ニューロンの変性・脱落が関わっていることが知られている。多くのパーキンソン病は、孤発性であるが、SNCA(α-シヌクレイン)などの遺伝子変異による家族性パーキンソン病も存在する。近年CHCHD2が、家族性パーキンソン病の原因遺伝子(PARK22)であり、孤発性パーキンソン病のリスク遺伝子となっていることが明らかにされた。しかし、そのパーキンソン病発症メカニズムに関しては未知な部分が多く、その疾患異常を改善する方法もまだわかっていない。
CHCHD2変異型<CK1<α-シヌクレイン/NEFLリン酸化<アグリソーム形成
研究グループは今回、CHCHD2の最も多い疾患原因変異である61番目のスレオニンがイソロイシンに変化したT61I変異によるパーキンソン病の発症機序を明らかにした。
具体的には、マウス由来神経芽細胞腫Neuro2a細胞を神経分化させた場合、CHCHD2野生型(CHCHD2WT)は、ミトコンドリアに局在しているが、T61I変異型(CHCHD2T61I)は、一度ミトコンドリアに入るものの、その後ミトコンドリア外に移動することがわかった。この間違って局在したCHCHD2T61Iは、CK1ε/δをリクルートすることで、そのキナーゼ活性によりα-シヌクレインと神経フィラメント構成因子(NEFL)のリン酸化を引き起こし、結果としてアグリソームの形成を誘導した。
CK1阻害剤によりリン酸化を大幅抑制、歩行異常の改善などをマウスで確認
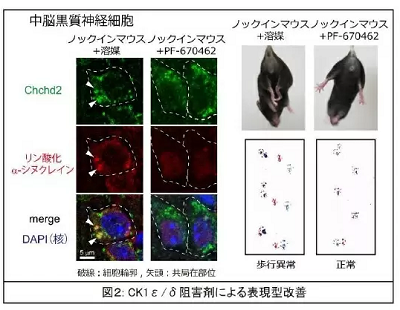
マウスを使った解析では、Chchd2T61Iノックインマウスおよびトランスジェニックマウスともに、ドーパミン作動性ニューロンにChchd2 T61I、CK1ε/δ、リン酸化α-シヌクレイン、リン酸化NEFLを含むアグリソームが形成され、神経変性特有の歩行異常、運動機能障害が見られた。同様のアグリソームは、死後PD患者の脳と患者由来iPS細胞から作製したドーパミン作動性神経細胞でも観察された。
そこで、CK1ε/δ阻害剤であるPF-670462で細胞やマウスを処理すると、α-シヌクレインとNEFLのリン酸化を大幅に抑制することができた。また、PF-670462は、ドーパミン作動性ニューロンの細胞死を抑制し、Chchd2T61I変異ノックインマウスのクラスピング(握り行動)や歩行異常などの神経変性表現型を改善させた。
パーキンソン病全体の疾患治療につながる可能性
今回の研究成果は、CK1ε/δがリン酸化α-シヌクレインを介して、CHCHD2T61I変異によるPDの病態に関与していることを示すものだ。このため今後CK1の阻害剤がCHCHD2T61I変異によるパーキンソン病の治療法候補の一つとして挙げられることが期待される。「リン酸化α-シヌクレインは他の家族性もしくは孤発性パーキンソン病にも広く関与することがわかっているため、今回の結果はパーキンソン病全体の疾患治療につながる可能性を含んでおり、今後のさらなる発展が期待される」と、研究グループは述べている。
▼関連リンク
・東京医科歯科大学 プレスリリース

