2本の相同染色体、区別し染色体スケールで決定することは困難
東京工業大学は8月7日、真核生物のゲノム配列決定において、両親由来の配列を区別し、染色体スケールでつながった配列をそれぞれ決定する、新しい情報解析手法の開発に成功したと発表した。この研究は、同大生命理工学院生命理工学系の大内俊大学院生(博士後期課程3年)、伊藤武彦教授、梶谷嶺助教(研究当時)らの研究グループによるもの。研究成果は、「Genome Biology」に掲載されている。
画像はリリースより
(詳細は▼関連リンクからご確認ください)
ロングリードシーケンス技術やHi-C法などDNA配列決定の技術革新により、少人数の研究グループでも、ゲノム中の染色体のほぼ全長の配列を決定できる時代が到来してきている。しかしながら、ヒトを含む2倍体生物のゲノム配列決定には依然として、以下のような課題がある。
1つ目の課題は、既存のHi-C法を用いた染色体スケールの配列決定ツールの結果にはエラーが含まれており、手動の修正が必要なことである。手動修正には大幅な時間がかかるうえに、作業担当者により結果が異なる再現性の問題もあり、自動化が望まれていた。
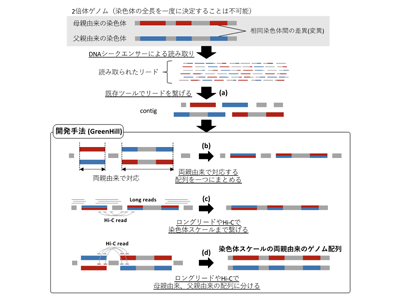
2つ目の課題は、2倍体生物のゲノムは、母親と父親からそれぞれ受け継いだ2本の相同染色体を持っているが、そのうち1本分しか決定できないことである。そこで、2本の相同染色体の違いを無視し、モザイク状につなぎ合わせた擬似的な1本のゲノム配列を決定する手法が長く用いられてきた。しかし近年、両親由来のゲノム配列(相同染色体間)で差異が大きい領域が表現型と関連する事例(ヒトの免疫型決定など)が報告されており、両親由来のゲノム配列を分けて解析することが重要であることがわかってきている。しかしながら、両親由来のゲノム配列を区別し、染色体スケールでつながった配列をそれぞれ決定することは、技術やコストの面から多くの問題が存在していた。
次世代・ロングリード・Hi-Cから染色体スケールの両親由来ゲノム配列構築プログラム開発
今回の研究で開発された解析手法では入力として、次世代シーケンサーの断片配列データを既存ツールでつなげた配列(contig)を用いる。解析ではまず、このcontig内の両親由来の配列から対応する配列を検出し、一つにまとめる。そのうえで、まとめた配列をロングリードやHi-Cのデータを用いて染色体スケールまでつなげる。最後に、最初にまとめた配列を二つに分け、ロングリードやHi-Cのデータを用いて、母親由来と父親由来の配列に分けることで、染色体スケールの両親由来のゲノム配列を構築する。
研究グループは、この手法を「GreenHill」というプログラムに実装し、公開した。さらにこの手法を実際に、線虫、牛、キンカチョウ、インコ、クロサイ、コチョウザメなどの各種生物に適用し、長くつながった両親由来の配列をそれぞれ既存の手法よりも高精度で決定できることを実証した。
がんゲノムの変異解析など幅広く応用可能
今回開発した手法は、2倍体生物のゲノム配列決定の真のゴールである「両親から引き継いだすべての染色体の全長の配列を決定すること」の達成に大きく貢献すると期待される。また、この開発手法は多くの生物種に対して高い性能を発揮しており、ゲノム配列決定プロジェクトを幅広く推進することも見込まれる。両親由来のゲノム配列を区別し、染色体スケールで決定することは、相同染色体間の大規模な変異の解析などの下流解析をする上で重要であり、がんゲノムの変異解析や植物の育種のためのゲノム解析など、幅広い分野への応用が期待される。
今回の研究により、配列データのみから両親由来の配列を染色体スケールで決定する方法の開発に成功した。「今後は、倍数性が高いなどの特徴を持つ複雑なゲノムの配列決定にも対応することで、より汎用性の高いゲノム配列決定手法の開発を目指す」と、研究グループは述べている。
▼関連リンク
・東京工業大学 プレスリリース


