希少がんの骨肉腫、p53遺伝子異常に続く腫瘍発症機構は?
長崎大学は8月9日、骨肉腫について、発症を制御する「がん遺伝子」「がん抑制遺伝子」間の新規な転写因子相互作用を解明したと発表した。この研究は、同大医歯薬学総合研究科分子硬組織生物学分野の伊藤公成教授、大谷昇平助教、伊達悠貴学振特別研究PD、口腔腫瘍治療学分野の大森景介助教らの研究グループによるもの。研究成果は、「Oncogene」に掲載されている。
画像はリリースより
(詳細は▼関連リンクからご確認ください)
骨肉腫は、主に小児をはじめ若年層に発症が多い希少がん。生存できても四肢の切除手術などにより、若くして失うものの影響が大きい。そのため、効果的な治療法の開発が急務だ。ヒトがんにおいて最も普遍的ながん抑制遺伝子p53遺伝子の不活性化が、骨肉腫において高頻度に観察される。しかし、希少がんであるため、国内外で骨肉腫の研究者人口は少なく、p53遺伝子の異常に続く腫瘍発症機構は、ほとんど解明されていない。
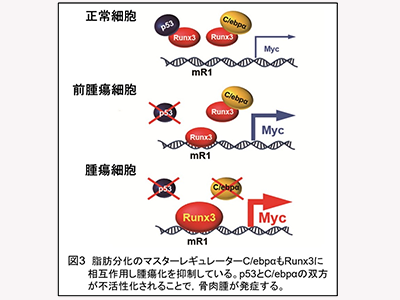
p53不活性化<フリーのRunx3がmR1に結合<Myc過剰発現<腫瘍化
骨芽細胞特異的p53遺伝子欠損マウス(OSマウス)は、100%に近い個体がヒト骨肉腫に酷似した腫瘍を発症する。そのため、ヒト骨肉腫の動物モデルとして使用されている。そこで、今回の研究では、骨肉腫発症の分子メカニズムの解明を目指し、このOSマウスを解析した。その結果、p53不活性化後に起こる腫瘍化プロセスの根本は、がん関連転写因子Runx3による、ゲノムDNA上の特定エレメントmR1を介した、強力ながん遺伝子c-Myc(Myc)の過剰発現であることが判明した。
そして、脂肪分化マスターレギュレーターとして知られる転写因子C/ebpαも、p53と同様に、Runx3をターゲットにしてタンパク質―タンパク質相互作用を示し、Runx3の機能を阻害することで、がん抑制遺伝子として機能していることが明らかになった。すなわち、p53非存在下でのRunx3によるmR1を介したMycの過剰発現は、肉腫発症の根本にある発がん機軸であると考えられる。
Runx3やmR1の阻害剤開発に期待
抗腫瘍効果を期待した創薬研究は数多く、その標的としてがん抑制遺伝子p53の賦活化やがん遺伝子Mycの機能阻害を目指した創薬が試みられてきた。しかし、両者とも多機能転写因子であるため、それらの機能を人為的に操作すると正常細胞への副作用が強いことから、いずれも奏功していないのが現状だ。そこで今回の一連の研究成果から、新たなる標的として両者を結びつけ、腫瘍の発症へと導く仲介役としてのRunx3の機能あるいはゲノム上の特定エレメントmR1が注目され、その効果的な阻害剤の開発が待たれる。
正常細胞では、Runx3の機能はp53やC/ebpαによって阻害されているため、Runx3の機能阻害による正常細胞への副作用は小さいことが期待される。また、Runx3はp53欠損膵がんにおいて悪性化(転移)促進因子として知られるようになった。同研究の成果は、骨肉腫のみならず、広くp53欠損型のヒトがんへの適用が期待される、と研究グループは述べている。
▼関連リンク
・長崎大学 プレスリリース

