CtBP2タンパク質の膵β細胞での役割は?
筑波大学は8月8日、肥満がインスリン分泌細胞の機能を低下させる仕組みを解明したと発表した。この研究は、同大医学医療系の島野仁教授と関谷元博准教授らの研究グループによるもの。研究成果は、「Cell Reports」に掲載されている。
画像はリリースより
(詳細は▼関連リンクからご確認ください)
肥満は糖尿病、脂肪肝、動脈硬化性疾患などのいわゆるメタボリックシンドローム関連の疾患群だけでなく、がん、精神疾患、免疫疾患など、あまり関連が認知されていないような多彩な疾患群の発症や病状の促進効果があることが知られている。そのため、肥満病態の中核的な因子を同定することはメタボリックシンドローム関連疾患だけでなく、領域横断的な疾患群の理解や治療法の開発につながる可能性を秘めている。
肥満において糖尿病は大きな問題となっているが、インスリン分泌細胞である膵β細胞に注目すると、経年的な肥満や代謝異常が膵β細胞の機能を低下させ、インスリン分泌が低下することが病態の一つの柱となっている。しかし、これまでの研究では、膵β細胞特異的なメカニズムの報告が多く、肥満に共通してみられるような病態に関する研究は少ない状況だった。研究グループは先行研究で、肥満病態が肝臓における代謝産物センサー分子CtBP2タンパク質の不活性化により惹起されること、また、これを活性化させると糖尿病や脂肪肝に治療効果を示すことを報告している。そこで今回、CtBP2の膵β細胞での役割を検証した。
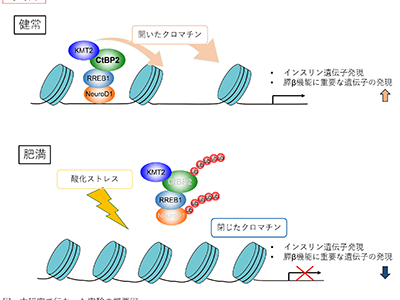
CtBP2が、NeuroD1の機能増強・膵β細胞機能維持・インスリン遺伝子発現に重要と判明
その結果、CtBP2は転写共因子であり、膵β細胞においては転写因子NeuroD1と結合して転写因子複合体を形成していることが明らかになった。
NeuroD1はインスリンや膵β細胞の機能維持に重要な遺伝子群の発現の調節を担っており、CtBP2はこのNeuroD1の機能を増強している。つまり、膵β細胞の機能維持やインスリン遺伝子発現に重要な役割を果たしていることが判明した。また、その遺伝子発現調節は、KMT2(酵素)などのヒストンメチル化などを介してクロマチン構造を調節することで行われていることも明らかになった。
肥満の酸化ストレスでCtBP2がユビキチン修飾を受けて分解され、発現量低下
さまざまな肥満モデルマウスにおいて、膵β細胞でのCtBP2は顕著に低下しており、ヒトの肥満者(脳死ドナーの検体)でもCBP2発現量が低下していることが観察されました。膵β細胞は酸化ストレスの消去能が他の細胞に比して弱いことが知られており、肥満によって生じる酸化ストレスによってCtBP2はユビキチン修飾を受けて分解されるため、発現量が低下するという仕組みが明らかになった。
長期肥満経過における膵β細胞機能低下と糖尿病の発症のプロセスにCtBP2が関連
さらに、膵β細胞特異的にCtBP2遺伝子を欠損させたマウスでは、胎生期から欠損させても生後に欠損させても、インスリン分泌低下型の糖尿病を示した。このことから、長期の肥満経過における膵β細胞機能低下と糖尿病の発症のプロセスは「肥満によってCtBP2量が低下」「インスリン分泌低下」「糖尿病発症」というCtBP2を中心とした分子メカニズムで説明できると考えられる。
肝臓のみならず膵β細胞も肥満におけるCtBP2の不活性化が病態の原因
今回の研究により、肝臓に続いて膵β細胞でも、肥満におけるCtBP2の不活性化が病態の原因であり、その活性化が治療効果を示すという結論に達した。「これは、肥満における膵β細胞の機能低下の新しい理解を得ただけでなく、今後、CtBP2が肥満の分子メカニズムの共通項として、臨床応用研究などの対象となることが期待される」と、研究グループは述べている。
▼関連リンク
・筑波大学 TSUKUBA JOURNAL


