ATMはすべての増殖細胞で機能するのに、変異でなぜ乳がんだけ増えるのか?
京都大学は2月9日、遺伝性乳がん(HBOC症候群)でBRCA1、BRCA2の次に頻度の高いATM遺伝子変異について、特定臓器(乳腺)の発がんを上昇させる分子機構を明らかにしたと発表した。この研究は、同大医学研究科の山田真太郎助教、Rifat Najnin大学院生、深圳大学の武田俊一教授、NYメモリアルスローンケタリングがん研究病院のScott Keeneyラボヘッドらの研究グループによるもの。研究成果は、「Cell Reports」にオンライン掲載されている。
画像はリリースより
(詳細は▼関連リンクからご確認ください)
ATMキナーゼは、ホモ欠損の場合、毛細血管拡張性運動失調症を発症するが、変異の保因者では乳がん発症が増加する。ATMの既知機能は、ゲノム切断時のP53活性化とDNA複製時に自然発生するゲノム切断を修復することである。これらは、すべての増殖細胞で機能し発がんを抑制するが故に、なぜ、保因者では特定の臓器(乳腺上皮)で発がんが増加するのか、その分子機構は不明だった。
ヒトゲノムは、生理的な状態でも多数の切断が各細胞で毎日発生する。自然発生するゲノム切断の原因の1つがDNA topoisomerase II(TOP2)の触媒反応の失敗である。切断は、染色体転座などの変異を起こして発がんを促進することが知られている。切断が早期転写応答に影響するかどうかは未知だった。
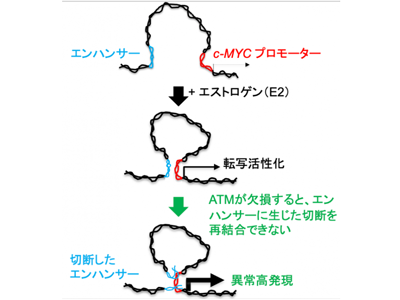
E2暴露により切断されたゲノム断片、ATMの欠損により修復が遅れる
研究グループは、まずヒト乳がん細胞とマウスをエストロゲン(E2)に短時間曝露し、ゲノム切断発生を免疫染色によって定量した。その結果、ATMが欠損すると、短時間曝露時に生じたゲノム切断が乳がん細胞とマウス乳腺上皮において1日近く修復されないまま残ることを発見した。切断発生には、エストロゲン受容体とTOP2の両方が必要だった。切断の再結合には、ATMと非常相同末端結合経路(G0/G1期に発生したゲノム切断の修復に必須)が両方共同する必要があることを確認した。
次に、ATMの基質とリン酸化サイトの決定を行った。データマイニングから、ATMの基質とそのリン酸化サイトをCtIPのThr847とThr859と予測した。その予測を遺伝学的手法で検証した。具体的には、野生型細胞とATM欠損(ATM-/-)細胞のリン酸化サイトに変異(CtIPT847A/T859A変異)をノックインし、ATM-/-、CtIPT847A/T859A、薬剤でATMを阻害したCtIPT847A/T859AがTOP2依存的ゲノム切断の修復についてよく似た表現型であることを示した。CtIPのDNA切断活性(既知)は、切断端に共有結合したTOP2を剥せる。この既知データと研究グループの遺伝学的解析データから、ATMの基質はCtIPと結論した。
ゲノム切断が効率よく修復できない細胞、エンハンサー活性化と早期転写応答に大きな変化
研究グループは、E2曝露時に生じたTOP2依存的ゲノム切断がE2応答性エンハンサーに発生したという仮説を立てた。早期転写応答中にエンハンサーが切断され、それがすぐに再結合されないと、早期転写に異常が生じる可能性がある。この可能性を検証するために、NET-CAGE法によって、E2曝露後の、エンハンサー活性と早期転写応答を継時的に調べた。ATM欠損細胞を含め、TOP2依存的ゲノム切断が効率よく修復できない細胞では、野生型細胞に比べ、エンハンサーの活性化と早期転写応答が大きく変化した。この結果は、(1)E2曝露への早期転写応答中にエンハンサーにゲノム切断が発生、(2)ゲノム切断がすぐに修復されないと早期転写応答が異常になる、の2点を示唆する。
E2によりc-MYCのエンハンサー切断、ATM欠損で再結合が遅れるとc-MYC発現が乳腺上皮で亢進
また、研究グループは、ヒトATM欠損乳がん細胞は野生型に比べ、E2曝露によるc-MYCの発現誘導がさらに3倍程度亢進し長く続くことを見つけた。そこでc-MYCのエンハンサーにTOP2依存的切断がE2曝露中に起こると考えた。ゲノム切断を検出する為に、H2AX抗原をクロマチン免疫沈降法で解析した。非相同末端結合欠損細胞をE2曝露中にエンハンサーが切断されることを確認した。さらにCRISPR/Cas9によって野生型乳がん細胞のエンハンサーにゲノム切断を発生させると、E2曝露中に、ATM欠損乳がん細胞にE2曝露した時のように、c-MYCの発現が亢進することを見つけた。以上の実験結果から、前段落の2つの結論、(1)と(2)が、c-MYCの、E2曝露への早期転写応答において正しいことを証明した。
研究グループは、E2を野生型マウスとATM欠損マウスに腹腔注射し、マウス乳腺上皮のc-MYC発現を組織免疫染色法によって解析した。E2注射をするとc-MYC+細胞は、野生型マウスでは4%から10%に増加したのに対し、ATM欠損マウスでは4%から30%にも増加した。そして、E2を毎日一回ずつ3日連続して腹腔注射すると、3日間に一回以上細胞分裂した細胞は、野生型マウスでは6%から14%に増加したのに対し、ATM欠損マウスでは6%から36%に増加した。この増殖細胞の増加は、E2と同時にエストロゲン受容体阻害剤もしくはc-MYC阻害剤も注射すると、完全に抑制された。以上の実験結果から、ATMが欠損すると、E2刺激時のc-MYC発現誘導がマウス乳腺上皮で亢進し、上皮が異常増殖すると結論した。
今回の研究では、ATM欠損が乳がん発症を促進する機序を解明した。早期転写応答中にエンハンサーにゲノム切断が発生することと、その切断再結合経路を解明したことによって、ゲノム切断修復経路と早期転写応答という、これまで無関係と考えられてきた2つの生化学反応のあいだに密接なクロストークが存在することを明らかにした。「このクロストークは、ATM欠損による前立腺がん発症と進行性小脳失調、およびHBOC症候群の発がん機序の解明に貢献する」と、研究グループは述べている。
▼関連リンク
・京都大学 最新の研究成果を知る


