心不全におけるCaイオン調節異常と病態形成の詳細は不明
京都大学は12月15日、転写抑制因子NRSFにより制御され不全心において発現亢進する遺伝子「GNAO1」の発現抑制が、複数のマウス心不全モデルにおいてその病態を改善すること、逆に心筋におけるGNAO1の過剰発現は心機能低下を引き起こすことを見出したと発表した。この研究は、京都大学大学院医学研究科循環器内科学の中川靖章助教、信州大学医学部循環器内科学教室の桑原宏一郎教授、同大医学部分子薬理学教室の山田充彦教授らの研究グループと、米ミシガン大学、順天堂大学などとの共同研究によるもの。研究成果は、「Circulation Research」オンライン版に掲載されている。
画像はリリースより
心疾患は、日本において悪性新生物に次いで死因の第2位であり、心不全は心疾患の中で最も高い割合を占める。超高齢社会の到来により、今後さらに心不全患者が増加することが予想されている。また、あらゆる心血管疾患の終末像である心不全は、近年の治療法の進歩にもかかわらず、いまだ予後不良の疾患症候群でもある。そのため、心不全の発症・進展のメカニズムを明らかにすることによる新規治療標的の同定、新規治療法開発は急務だ。
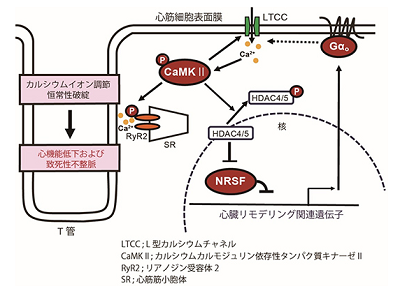
心不全は、心筋の収縮や弛緩の障害を基盤とする予後不良の症候群であり、その病態に心筋細胞内カルシウムイオン調節の異常が深く関与することが知られている。心筋細胞内カルシウムイオン調節においては細胞外から細胞内へカルシウムイオンを透過させるL型カルシウムチャネル(LTCC)と、細胞内に存在しカルシウムイオンを蓄えている心筋筋小胞体(SR)からのリアノジン受容体2(RyR2)を介したカルシウムイオン動員が重要な役割を果たす。正常な心筋の収縮には、心筋に存在するT管と呼ばれる膜構造におけるLTCCとSRとの連関が重要であることが知られている。
心不全ではこうしたカルシウムイオン動員機構に異常が生じている。T管でのLTCC活性が低下し、それ以外の表面細胞膜においてLTCC活性が上昇することや、カルシウムカルモジュリン依存性タンパク質キナーゼII (CaMKII)などの病的なカルシウムシグナルが活性化しSR上のRyR2のリン酸化を引き起こし、その正常機能を阻害していることなどが知られている。しかし、これらの病態形成に関与する詳細なメカニズムは明らかではなく、その解明による新規治療薬開発が望まれている。
NRSF制御のGNAO1が不全心筋で発現亢進、GNAO1抑制でマウス心不全が改善
研究グループは、心不全の心臓において発現が亢進する遺伝子群に注目し、それら遺伝子の発現を制御する因子として転写抑制因子neuron-restrictive silencer factor(NRSF)の意義を以前に見出していた。そして今回、心筋特異的NRSFノックアウトマウスおよび心筋特異的優勢抑制変異型NRSF過剰発現マウスの解析を通じて、NRSFにより制御される遺伝子であるGNAO1が不全心筋において発現亢進していることを見出した。
また、ノックアウトマウスによるGNAO1の発現抑制が、上記マウスを含む複数の心不全モデルマウスにおいてその病態を改善すること、一方、心筋におけるGNAO1の過剰発現は心機能低下を引き起こすことも明らかになった。GNAO1によりコードされるタンパク質であるGαoの病的ストレスによる心筋での発現亢進が、心機能低下および心不全の発症・進展に重要な役割を果たしていることも世界で初めて明らかになった。
GNAO1/Gαo発現亢進<LTCC活性上昇<病的Caシグナル活性化<Caイオン恒常性破綻
さらに、そのメカニズムとして、Gαoの発現亢進が心筋表面細胞膜におけるL型カルシウムチャネルの局在活性を上昇させることで心筋細胞内での病的カルシウムシグナル経路の活性化とそれに引き続くカルシウムイオン調節の恒常性破綻をきたすことであることがわかった。
同研究は心不全の発症・進展の新たなメカニズムの解明であり、超高齢社会の到来により急増する心不全の病態の理解をさらに進めるものとなる。「また、Gαoを標的とした新しい心不全治療薬の開発にもつながる可能性があり、いまだ予後不良の疾患症候群である心不全に対する新しい治療薬の開発に役立つことが期待される」と、研究グループは述べている。
▼関連リンク
・京都大学 最新の研究成果を知る


