

ALS患者で起こる糖代謝異常の原因は不明だった
名古屋大学は7月30日、難治神経変性疾患のひとつである筋萎縮性側索硬化症(ALS)は、病初期からインスリン分泌能が低下することを見つけ、ALS患者の膵臓にあるβ細胞の核においてALSの病態分子であるTAR DNA-binding protein of 43kDa(TDP-43)が喪失していることを見出し、さらに、TDP-43が電位依存性Caチャネル(CaV1.2)の転写活性を調整することによってインスリン分泌を制御していることを明らかにしたと発表した。この研究は、同大学大学院医学系研究科神経内科学の勝野雅央教授、愛知医科大学の祖父江元理事長、名古屋大学大学院医学系研究科糖尿病・内分泌内科学の有馬寛教授、同医学部附属病院脳神経内科の荒木邦彦医員(筆頭著者)らの研究グループによるもの。研究成果は、「Journal of Clinical Investigation」電子版に7月30日付で掲載された。
画像はリリースより
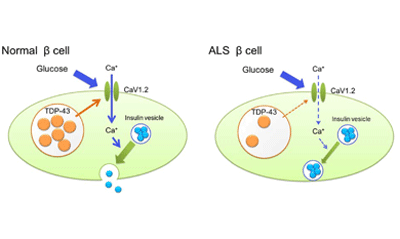
ALSは、運動ニューロンの死滅を原因とする急速進行性の筋萎縮により、発症後、数年で死に至る神経変性疾患。2006年に孤発性ALSの病態分子としてTDP-43が発見された。TDP-43は、本来、核内に存在するが、ALS患者の運動ニューロンでは核内のTDP-43は消失し、核の外側(細胞質)に異常なタンパク質の塊である封入体を形成することから、TDP-43の機能異常(Loss of function)と TDP-43凝集体の細胞毒性(gain of toxic)の両者が病態に影響すると考えられている。勝野教授らの研究チームは、これまでTDP-43の機能異常に注目し、ニューロンの核からTDP-43が消失すると細胞が弱ることを明らかにしてきたが、TDP-43喪失からニューロン変性にいたるメカニズムは不明であった。また、ALS患者は、しばしば血糖が上がりやすくなること(糖代謝異常)が知られているが、その原因は不明であった。
TDP-43が膵β細胞でCaV1.2の遺伝子発現を制御
研究グループは今回、ALSの神経系以外の全身症状に着目をし、ALS患者でブドウ糖負荷試験を行った結果、インスリンの分泌能が低下していた。また、患者の膵臓組織(死後に病理解剖で得られたもの)において、インスリンを産生するβ細胞の核内でTDP-43が喪失していることを発見した。そこで、β 細胞のTDP-43喪失がインスリン分泌能低下を引き起こすという仮説を立て、β 細胞株(MIN6 細胞)を用いて TDP-43 をノックダウンした。この細胞を用いたグルコース負荷試験、および特殊な顕微鏡による視覚的評価により早期相のインスリン分泌低下を立証。また、TDP-43ノックダウン時のMIN6細胞の網羅的な遺伝子解析からCaV1.2の低下を認め、TDP-43がCaV1.2の遺伝子プロモーター領域に結合し、転写活性を上昇させることを示した。次に、膵臓特異的TDP-43ノックアウトマウスを作製し、種々の解析を行った結果、β細胞におけるCaV1.2の発現低下とインスリン分泌能の低下を確認した。
今回の研究では、運動ニューロンにのみ変性が起きると思われていたALSにおいて、糖代謝異常が生じ、膵臓にまで病気が広がっていることが証明された。これは、ALSにおける運動ニューロン以外でのTDP-43の機能喪失を示した重要な知見。「将来的に膵臓におけるインスリン分泌能がALSの病態を反映するバイオマーカーとなることが期待される。また、インスリン分泌機構におけるTDP-43の役割が明らかとなったことから、糖尿病の新たなメカニズム解明にもつながると考えられる」と、研究グループは述べている。
▼関連リンク
・名古屋大学大学院医学系・医学部医学科 研究トピックス


