両親由来のゲノム配列を「分けて」解析したい
東京工業大学は4月22日、真核生物のゲノム配列決定において、両親由来の配列を区別し、高精度にそれぞれを決定する新しい情報解析手法の開発に成功したと発表した。この研究は、同大生命理工学院生命理工学系の梶谷嶺助教、吉村大大学院生(研究当時)、奥野未来研究員、伊藤武彦教授らと、国立遺伝学研究所の豊田敦特任教授、小原雄治特任教授、東京大学の窪川かおる特任教授らが共同で行ったもの。研究成果は、「Nature Communications」に4月12日付で掲載された。
画像はリリースより
ヒトなど真核生物のゲノム情報は、両親から受け継いだ情報を持ち合わせているが、現状ではこの差異を無視して、両親由来のゲノム情報全体をモザイク状につなぎ合わせることで、ゲノム配列を解読する手法が一般的に用いられている。一方で、近年、両親由来のゲノム配列(相同染色体)間で差異が大きい領域が存在し、これがさまざまな表現型(昆虫の体の紋様や性決定、ヒトの免疫型決定や疾患など)と関連している事例が報告されている。そのため、両親由来のゲノム配列を「分けて」解析することの重要性が認識されるようになり、簡便に両親由来の配列を区別して解析できる手法が求められていた。
特殊な装置や前処理を必要とせず高精度に決定
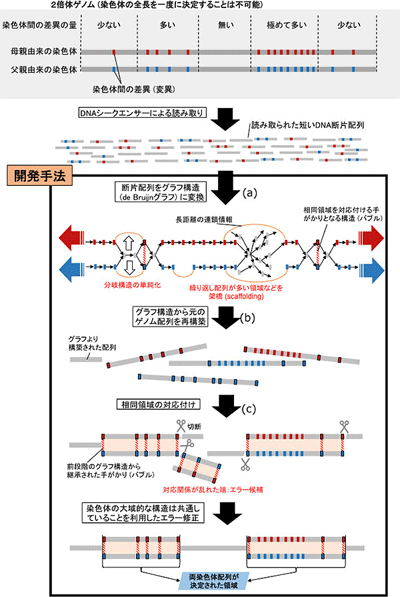
今回研究グループが開発した解析手法は、次世代シークエンサーの大規模な断片配列データ(ショートリード)の精度の高さを生かして、まず、大量の断片配列内に存在する一塩基の違いをも区別できるグラフ構造(ノードとエッジで表現されるデータの集合体)に変換。次に、そのグラフ構造をショートリード間のペア情報を用いて単純化することで、より長く繋がったゲノム配列を再構築。さらに、相同染色体との対応付けを配列の類似情報から行い、エラー修正などを行った上で最終的な配列を導き出すというもの。
研究グループは、この手法を「Platanus-allee(プラタナスアリー)」というプログラムに実装し、インターネットで公開している。実際に、この手法を、線虫、シロオビアゲハ、ナメクジウオ、サクラ、ヒトなどの各種生物に適用したところ、その実効性を証明できたという。
今回開発された解析手法で、相同染色体間の複雑な変異情報が網羅的に収集可能となる。これにより変異が蓄積したゲノム領域との関連が疑われている種分化、多様性維持、免疫など重要な生命現象の解明が進むと考えられる。また、「究極的には、この研究成果を用いて、ヒトを含む「2倍体生物が2セットの少し異なるゲノムを維持することで何を得たのか?」という生命現象に対する根源的な問いへの理解が深化することが期待される」と、研究グループは述べている。
▼関連リンク
・東京工業大学 ニュースリリース


