病原性Th17細胞を制御する詳細な仕組みについて研究
大阪大学は2月7日、遺伝子発現の制御分子であるSatb1に着目し、IL-17サイトカインを産生するヘルパーT細胞(Th17細胞)が病気を引き起こす仕組みを、ヒトの自己免疫疾患である多発性硬化症のマウスモデルを用いて明らかにしたと発表した。この研究は、同大学の安田圭子医員(医学部附属病院、医学系研究科腎臓内科学)、坂口志文特任教授(免疫学フロンティア研究センター)および京都大学の廣田圭司准教授(ウイルス・再生医科学研究所兼大阪大学招へい准教授)らの研究グループによるもの。研究成果は、英科学誌「Nature Communications」(オンライン)に2月1日付で掲載された。
画像はリリースより
Th17細胞は、体の正常な機能を保つ(腸管での生体恒常性の維持、真菌感染に対する防御)働きを持つ一方で、自己免疫疾患(多発性硬化症、乾癬、関節リウマチなど)において自分の体を攻撃して病気を起こす。Th17細胞が病原性を獲得するには、IL-17を産生するようになった後に、IL-1βやIL-23などの炎症性のサイトカインの暴露を受けることが重要であることが、これまでに知られている。しかし、生体内でIL-17を産生するようになった細胞を、生きた状態で回収して性質を調べることが困難であったため、病原性Th17細胞を制御する詳細な仕組みについては、これまで明らかになっていなかった。
転写因子「Satb1」を欠損すると自己免疫疾患になりにくいと判明
今回、坂口特任教授らの研究グループは、転写因子「Satb1」に着目。Satb1は、T細胞の分化に重要であることは分かっていたが、生体内でサイトカインを産生するヘルパーT細胞でのSatb1の役割についてはこれまで不明だった。そこで、Th17細胞がIL-17産生後、病原性を獲得する過程におけるSatb1の役割を調べた。
これまでに、廣田准教授らの研究グループは、IL-17を産生したことがある細胞を蛍光色素で標識するリポーターマウスの作製に成功し、研究を進めていた。今回研究グループは、IL-17産生リポーターマウスを用いて、IL-17を産生後にSatb1の発現を欠損させ、同時にそれらの細胞を蛍光色素で標識するマウスを作製。このマウスを用いて、Satb1を欠損するTh17細胞をマウス生体から生きた状態で回収することに成功した。
作製したマウスを用いて解析をした結果、ナイーブCD4+T細胞からTh17細胞に分化する過程では、Satb1の有無による影響は見られず、また、腸管に存在する、病気を起こさないタイプのTh17細胞はSatb1を欠損しても機能に影響しないと分かった。一方で、Th17細胞においてSatb1を欠損するマウスは、多発性硬化症マウスモデル(EAE:実験的自己免疫性脳脊髄炎)で、病気になりにくいことを見出した。
自己免疫疾患に対する新しい治療法の開発に期待
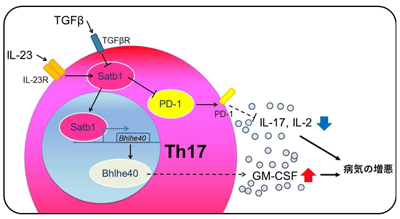
そのマウスの脳脊髄や炎症を起こしているリンパ節に含まれるTh17細胞について解析をしたところ、Th17細胞でのSatb1の欠損は、EAEマウスにおいて、病気の発症に必須のサイトカイン「GM-CSF」の産生を抑え、さらに免疫チェックポイント分子「PD-1」の発現を上昇させることで病気を軽減させることが分かった。また、Th17細胞がIL-23に暴露されるとSatb1の発現が亢進し、逆にTGF-βでは発現が抑えられたことから、Th17細胞でのSatb1の発現は、細胞をとりまく環境中に存在するサイトカインの影響を受けて変化し、その結果としてTh17細胞の病原性を決定することが分かった。
さらに詳細に解析を行なった結果、転写因子「Bhlhe40」の発現を調節する遺伝子領域にSatb1が転写因子として直接作用しBhlhe40の発現を亢進させること、さらにBhlhe40の作用によって、GM-CSFの産生を増加させることが明らかとなった。加えて、別の経路として、EAEの脳脊髄に浸潤しているTh17細胞において、Satb1の欠損はPD-1の発現を亢進する方向に働き、IL-17やIL-2といったサイトカインの産生を抑制し、結果としてEAEの病勢を抑えることが分かった。
今回の研究結果から、Th17細胞でのSatb1の発現を抑えることを治療の標的とすることで、多発性硬化症のような自己免疫疾患に対する新しい治療法の開発に結び付くことが期待される、と研究グループは述べている。
▼関連リンク
・大阪大学 研究情報

