家族性脊髄小脳失調症の原因とされる「IP3受容体I型」
理化学研究所は11月13日、家族性脊髄小脳失調症の原因遺伝子であるカルシウムチャネルIP3受容体(イノシトール三リン酸受容体)I型の点突然変異の影響を網羅的に解析し、変異によりIP3受容体I型が機能不全に陥る分子機構を解明したと発表した。この研究は、理研脳神経科学研究センター発生神経生物研究チームの安東英明研究員(研究当時)、御子柴克彦チームリーダーらの研究チームによるもの。研究成果は「Proceedings of the National Academy of Sciences of the United States of America(PNAS)」の掲載に先立ち、オンライン版に掲載されている。
画像はリリースより
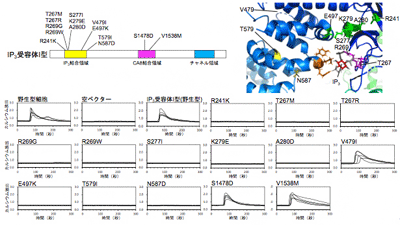
家族性脊髄小脳失調症は、その原因遺伝子により40以上の型に分類されているが、そのうち15/16型および29型の原因遺伝子が、細胞内のカルシウムチャネルであるIP3受容体I型であることが近年の臨床研究で明らかになっている。IP3受容体には3種類の遺伝子(I型、II型、III型)があり、そのうちIP3受容体I型は、小脳の神経細胞に多く発現し、脳機能に重要な役割を果たしている。家族性脊髄小脳失調症の15/16型が主にIP3受容体I型の大規模な欠失突然変異が原因であるのに対し、29型はIP3受容体I型の点突然変異が原因だ。現在までに、家族性脊髄小脳失調症で約20か所のIP3受容体I型の点突然変異が見つかっている。
しかし、これらの変異がIP3受容体I型の機能にどのような影響を及ぼすかについては、ほとんど解明されていなかった。これは、ほぼ全ての細胞において、3種類のIP3受容体のいずれかが発現しており、正常なIP3受容体活性が存在するために、ヒト由来の細胞を用いてIP3受容体の変異体の活性を測定することがこれまで困難だったためだという。
IP3受容体I型のミスセンス変異でカルシウムシグナルに異常
そこで研究チームは、ゲノム編集技術を用いて3種類のIP3受容体遺伝子が全て欠失したヒト細胞株を新たに樹立。家族性脊髄小脳失調症で見つかった点突然変異を持つIP3受容体I型変異体の活性を網羅的に解析した。
その結果、IP3受容体I型のミスセンス変異により、その活性化機構や、結合タンパク質による制御が正常に機能しなくなり、細胞のカルシウムシグナルに異常をきたしたりすることが判明。これにより神経細胞内のカルシウム動態の異常が脊髄小脳失調症の発症に関わることを考えられるという。
研究チームは、2017年にIP3受容体I型のX線結晶構造の解明に成功しており、「今後、立体構造情報に基づきIP3受容体I型の阻害剤や活性化剤を探索することにより、家族性脊髄小脳失調症の治療のための創薬につながる可能性がある」と述べている。
▼関連リンク
・理化学研究所 プレスリリース


