

国際共同研究による全ゲノムシーケンス解析で4,600万個を超える変異・異常を同定
理化学研究所は2月6日、がん38種2,800例以上の全ゲノムシーケンス解析を行い、4,600万個を超える変異・異常を同定し、その特徴を明らかにしたと発表した。これは、同研究所生命医科学研究センターがんゲノム研究チームの中川英刀チームリーダー、東京大学医科学研究所健康医療データサイエンス分野の井元清哉教授らを含む国際共同研究グループ(PCAWG)によるもの。研究成果は、科学雑誌「Nature」(特集号)の掲載に先立ち、オンライン版に掲載されている。
画像はリリースより
がんは、ゲノムの変異や異常が蓄積することで発症・進行する「ゲノムの病気」である。現在、世界中でがんの網羅的なゲノム解析やゲノム情報に基づく薬の開発・個別化医療(がんゲノム医療)が精力的に行われている。世界最大規模のがんゲノム国際共同体「国際がんゲノムコンソーシアム」、米国主導の大規模ながんゲノム解析プロジェクト「The Cancer Genome Atlas」などにおける主ながんゲノム研究では、全ゲノムの約1~2%に相当するタンパク質をコードする領域(エクソーム)を主に解析し、そのデータが蓄積されてきた。しかし、近年の次世代シーケンサー技術の急速な進展、情報解析技術やITハード面の技術革新に伴い、ヒトの約30億塩基対の情報からなる全ゲノムでのシーケンス解析が、容易かつ安価に行えるようになった。このような背景から、今後は、全ゲノムシーケンス解析が研究のみならず、がんゲノム医療といった診断や個別化医療においても、重要な解析手法になると予測されている。
ほとんど全てのがんでの変異・異常が検出可能に
研究は2014年に始まり、37か国1,300人以上の科学者、ITエンジニア、臨床家が参加。世界10か所にあるゲノム解析センターのスーパーコンピューターをデータセンターとし、アマゾンやマイクロソフトのクラウドサーバーとも仮想マシーン(VM)で接続することでこれらを同期させ、統一された計算空間を構築した。データセンターとして、東京大学医科学研究所のスーパーコンピューター「SHIROKANE」も使われた。研究に参加する世界中の研究者は、これらのデータに容易にアクセスでき、計算資源を最大活用できる。
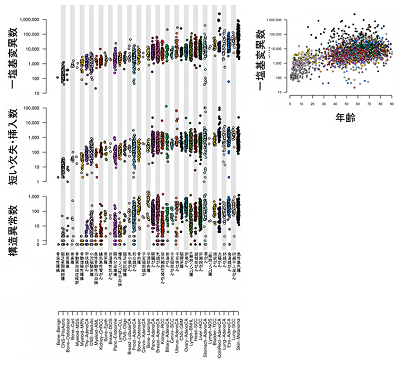
今回のがん全ゲノムの大規模解析では、ほとんど全てのがんでの変異・異常が検出可能。解析の結果、4,400万個の一塩基変異、240万個の短い欠失・挿入、29万個の構造異常、約8,000個のミトコンドリアゲノム異常、テロメア/TERT遺伝子異常など、合計で4,600万個を超える変異・異常が同定され、それらのさまざまな特徴も明らかになった。例えば、がん体細胞の一塩基変異数は、がんの診断年齢が高いほど多い傾向にあることもわかった。こうして、これまでで最も網羅的かつ詳細ながんゲノムマップが作成された。
今後、DNAシーケンス解析技術の革新に伴うコストの低下により、全ゲノムシーケンス解析が、研究分野のみならず、がんの診断や個別化医療の分野においても、標準的なゲノム解析手法になると予測されている。「PCAWGでの全ゲノムデータや開発した解析手法は、世界中で公開されている。データセットは、次世代のがんゲノム医療および研究のデータ基盤になると期待できる」と、研究グループは述べている。
▼関連リンク
・理化学研究所 研究成果


